Aidez-nous
Qu’est-ce que l’HPN ?
L’hémoglobinurie paroxystique nocturne (HPN), ou syndrome de Marchiafava-Micheli, est une maladie rare dont les premiers cas ont été décrits à la fin du XIXe siècle.
Selon une étude publiée le 10 mai 2024 intitulée « Algorithme d’identification des patients atteints d’Hémoglobinurie paroxystique nocturne (HPN) à partir des données du PMSI », la prévalence réelle de l’HPN en France ne peut être déterminée avec précision. Cependant, elle est estimée entre 1/70 000 et 1/80 000 habitants, soit entre 850 et 1 000 patients. Une analyse fondée sur le Système National des Données de Santé (SNDS) permettrait d’obtenir des données plus précises.
Le nombre de patients souffrant de cette maladie est aujourd’hui sous-estimé car elle est encore difficile à diagnostiquer.
Il s’agit d’une maladie acquise durant la vie. Cette maladie n’est donc pas transmissible à la descendance. Elle n’est pas contagieuse non plus.
L’HPN se caractérise par une destruction des globules rouges, appelée hémolyse. Cette hémolyse a lieu dans les vaisseaux, d’où la libération d’hémoglobine dans les urines qui deviennent foncées voir couleur porto. Cette destruction se fait de façon permanente avec des exacerbations poussées souvent déclenchées par des infections. L’hémolyse est la principale cause des différents symptômes observés chez les personnes atteintes d’HPN.
L’HPN touche le plus souvent de jeunes adultes, aussi bien les hommes que les femmes. Elle peut survenir chez les sujets âgés mais est exceptionnelle avant 15 ans. Le seul facteur favorisant sa survenue est l’aplasie médullaire (et beaucoup plus rarement d’autres pathologies hématologiques).
Quelle est son origine ?
L’HPN est une maladie qui entraîne la formation de globules rouges anormaux, appelés globules rouges HPN (GR HPN). Ces globules rouges HPN ont perdu (entre autre) leur système de protection contre l’attaque d’un système de défense naturel de l’organisme appelé le « complément » et vont donc être détruits. C’est cette destruction que l’on appelle hémolyse.
Le défaut d’expression de ces molécules protectrices à la surface des cellules du sang est dû à la survenue d’une mutation du gène PIGA dans une ou plusieurs des cellules souches de la moelle. Ces cellules présentes dans la moelle osseuse ont pour fonction de produire les globules rouges (GR), les globules blancs (GB) et les plaquettes présents dans le sang. Ces mutations sont acquises (elles apparaissent au cours de la vie) et ne sont pas présentes dans les autres cellules de l’organisme, notamment les gamètes (ovocytes et spermatozoïdes). Le déficit concerne toutes les cellules du sang mais il s’exprime essentiellement sur les globules rouges.
La coexistence de cellules souches mutées et non mutées explique que les GR HPN cohabitent avec des GR normaux.
Les GR HPN sont plus fragiles et peuvent être détruits par « le complément ».
Le « complément » fait partie de notre système de défense immunitaire et permet normalement de détruire les organismes étrangers. Mais dans notre cas, le système de protection des GR étant absent, le « complément » se fixe sur les GR et les détruit.
Le nombre de GR circulant dans le sang peut ainsi devenir insuffisant et entraîner une anémie importante.
L’HPN est étroitement lié à l’aplasie médullaire immunologique : on pense que son développement est favorisé par l’attaque immunologique des cellules souches observée dans l’aplasie. Ainsi au diagnostic d’aplasie médullaire, 40 à 50% des patients ont un clone HPN mais le plus souvent de petite taille et sans conséquence. Au total, 30% des patients avec un HPN hémolytique ont un antécédent d’aplasie médullaire traitée et 20 à 30% des patients avec HPN hémolytique développeront une aplasie médullaire dans les mois ou les années qui suivent le diagnostic.

Quels sont les symptômes ?
Le type, la fréquence et la sévérité des symptômes peuvent varier considérablement en fonction des patients. La plupart des symptômes sont peu spécifiques et apparaissent progressivement (fatigue, douleurs abdominales, essoufflement, troubles de l’érection) ce qui retarde le diagnostic si les anomalies hématologiques associées ne sont pas mises en évidence.
Principaux symptômes
L’anémie
Très souvent, l’HPN se manifeste par des épisodes plus ou moins fréquents d’anémie due au manque de globules rouges alors en quantité insuffisante pour apporter l’oxygène aux tissus. Les signes d’anémie sont généralement : pâleur de la peau, fatigue et parfois essoufflement à l’effort ou difficultés à respirer (dyspnée).
L’hémoglobinurie
L’hémoglobinurie est caractérisée par une coloration foncée des urines plus marquée le matin. L’hémoglobine se transforme en pigment brun-jaune (bilirubine) et est éliminée dans les urines qui deviennent plus foncées voire dans certains cas franchement brunes (couleur « porto »).
Jaunisse (ictère)
La bilirubine va également colorer la peau, entraînant l’apparition d’une jaunisse ou ictère.
La fatigue
La fatigue est l’un des symptômes les plus fréquents de l’HPN et sûrement l’un des plus invalidants, car elle est souvent assez intense pour limiter la réalisation des activités quotidiennes des patients, ralentir leur vie sociale et provoquer des épisodes de somnolence dans la journée.
Autres symptômes
D’autres symptômes peuvent apparaître en fonction de l’évolution de l’HPN. Ils surviennent par poussées et peuvent coïncider avec certains événements de la vie (infections virales, vaccination, opération chirurgicale et de façon générale toute situation où une inflammation survient).
Il peut s’agir notamment :
- de difficultés à avaler (dysphagie)
- de douleurs à l’abdomen ou au thorax ou au thorax ou de maux de tête
- de formation de caillots sanguins pouvant entraîner des thromboses (dont les symptômes varient selon leur localisation)
- de troubles de la fonction érectile
- de troubles rénaux en cas d’hémolyse sévère

Quelle évolution ?
L’évolution de l’HPN varie en fonction des personnes atteintes. Le principal facteur prédictif de la survenue de manifestations liées à l’HPN est la taille du clone (% des globules blancs HPN/ normaux). En deçà de 10%, il n’y a pas de manifestations. Entre 10 et 50% elles sont rares. Au deçà de 50% le risque est beaucoup plus élevé mais l’hémolyse reste variable d’un individu à l’autre. La sévérité clinique est corrélée à l’importance de l’hémolyse dont découle la profondeur de l’anémie, la fatigue, les manifestations de dystonie (douleurs abdominales, dysphagie, troubles de l’érection notamment) et le risque de thrombose ; Chaque cas est donc différent.
La formation de caillots (thromboses) est la complication la plus grave de l’HPN. Cela peut survenir dans n’importe quelle partie du corps mais certaines thromboses rares sont inhabituellement fréquentes : c’est le cas de la thrombose de certaines veines hépatiques, appelée syndrome de Budd Chiari et des thromboses des veines cérébrales.
En présence d’une aplasie médullaire, l’anémie sera également plus prononcée car en plus de la destruction des globules rouges, la production diminue. Il existe également une baisse de la production de certains globules blanc (neutrophiles) et des plaquettes qui peuvent être responsables d’infections sévères et d’hémorragie. Ces complications peuvent être fatales.
Plus rarement, une leucémie aiguë ou un syndrome myélodysplasiques (formes de cancer du sang) peuvent survenir au cours de l’évolution (8 à 15% après 20 ans de suivi).
Plus rarement, après plusieurs années (5 à 20 ans), chez à 3% des patients le clone HPN peut diminuer en deçà de 10% voir disparaitre et une rémission clinique complète spontanée est possible. Chez ces patients, les traitements sont donc arrêtés.
Quel diagnostic ?
Le diagnostic de l’HPN se fait par cytométrie de flux et ne nécessite qu’une prise de sang.
Cette méthode permet à partir d’un prélèvement de sang de déterminer la présence des globules blancs HPN (qui ont perdu l’expression de certaines protéines à leur surface) et d’en évaluer la quantité (taille du clone). On peut également mesurer la taille du clone sur les globules rouges mais c’est moins fiable du fait de leur destruction et ce dernier, sans traitement, est sous-évalué. On peut détecter des clones de taille très variable de 0.1% à 99,9%. On parle d’HPN quand le clone est supérieur à 10% et qu’il s’associe à des manifestations hémolytiques ou thrombotiques.
La cytométrie de flux n’est pratiquée que par certains laboratoires d’analyses, et il est préférable de réaliser ces analyses dans les laboratoires hospitaliers qui appliquent des règles de diagnostic et de rendu des résultats standardisées.
Un myélogramme (ponction de moelle osseuse sous anesthésie locale au niveau du sternum ou du bassin) avec une analyse du caryotype médullaire est recommandé au moment du diagnostic pour rechercher une autre pathologie associée. En cas de suspicion d’aplasie médullaire (plaquettes et neutrophiles très bas), une biopsie de moelle est également réalisée sous anesthésie locale pour confirmer le diagnostic.
Enfin, si la présence d’une thrombose (caillot) est suspectée, des examens d’imagerie peuvent être prescrits, comme un scanner, IRM ou échographie permettant de vérifier ou d’infirmer sa présence.
Quels sont les traitements ?
Il existe différents types de traitements médicamenteux de l’HPN. Ils sont efficaces, mais, aucun ne permet une guérison complète de l’HPN. Ils apportent toutefois un réel soulagement et une sécurité de vie. Le choix de ce traitement dépend de chaque patient et des signes de sa maladie.
Si une aplasie médullaire sévère ou un syndrome myélodysplasique est associée, une allogreffe de cellules souches hématopoïétiques (ou greffe de moelle) peut être proposée : elle permet de guérir de toutes ces pathologies. Ce traitement n’est plus proposé aux patients ayant une HPN isolée ou associée à une aplasie non symptomatique du fait des risques de complications sévères liés à la greffe et de l’existence de traitements alternatifs efficaces. Chez les sujets de moins de 40 ans ayant un donneur compatible dans la fratrie c’est le traitement de référence en cas d’aplasie sévères, pour les autres un traitement immunosuppresseur est proposé en 1ère intention et la greffe n’est proposée qu’en cas de rechute, d’absence de réponse ou d’évolution vers une maladie tumorale de la moelle.
Les soins de support
Ils permettent de prendre en charge différents symptômes.
La transfusion sanguine est envisagée, en cas d’anémie sévère ou mal tolérée, se traduisant par des difficultés à respirer et une grande faiblesse. Il s’agit d’injecter au patient des globules rouges d’un donneur, pour maintenir constant le niveau de globules rouges. En complément, du fer et de l’acide folique peuvent être prescrits car leur perte est importante lors de l’hémolyse.
Les anticoagulants fluidifiant le sang, comme l’héparine, peuvent être prescrits en cas thrombose ou de situation à haut risque (chirurgie, inflammation importante, voyage en avion, grossesse …). Ils ne sont plus prescrits en prophylaxie au long cours car ils n’empêchent pas la thrombose liée à l’HPN contrairement aux inhibiteurs du complément.
Des antibiotiques peuvent être prescrits en cas d’infections.
Question : les spasmes de l’estomac peuvent être sensibles à des dérivés nitrés comme la trinitrine.
Les inhibiteurs du complément qui sont de deux types :
1) Les inhibiteurs du complément de la partie terminale : L’anti-C5 disponible depuis 2007.
Retrouvez le replay de la 6ème journée nationale Médecins/Patient (juin 2023) sur les nouveaux inhibiteurs terminaux dans l’HPN en cliquant ici.
L’éculizumab, anticorps monoclonal humanisé anti-C5, a été pendant de nombreuses années le seul représentant commercialisé de cette classe. C’est un inhibiteur du complément qui empêche la destruction des globules rouges (hémolyse). Il est administré par perfusion veineuse toutes les 2 semaines.
Il permet de prévenir les complications de l’HPN : l’anémie et les thromboses, il améliore la qualité de vie des patients qui, ainsi traités, peuvent souvent reprendre activités professionnelles et sociales. Il permet ainsi de réduire significativement la fréquence des transfusions et même parfois de les supprimer. Ce médicament a permis d’améliorer la survie des patients qui était de 50% à 20 ans du diagnostic avant et est désormais proche de la normale pour l’âge.
Depuis mars 2022, le ravulizumab est commercialisé en France. Il agit de la même façon que l’éculizumab, mais il a une durée d’action plus longue, et réduit les hémolyses déclenchées par des infections et cela permet d’espacer les perfusions qui sont nécessaires seulement toutes les 8 semaines. Ce qui a un impact important sur la qualité de vie des patients.
Ces deux médicaments augmentent le risque d’infections à méningocoque, une bactérie dont le corps se défend grâce au complément. Ces infections pouvant être très graves (mortelles ou laissant des séquelles sévères), la vaccination contre le méningocoque est obligatoire avant le traitement puis régulièrement tant que le traitement est poursuivi. En France, on associe systématiquement une prophylaxie par antibiotique (pénicilline ou autre si allergie). Une consultation aux urgences en cas de fièvre est impérative.
2) Les inhibiteurs du complément de la partie proximale : Les inhibiteurs dits proximaux qui agissent plus en amont des anti-C5 et sont efficaces chez les patients qui répondent mal aux inhibiteurs du C5.
Chez certains patients des fragments de compléments se déposent sur les GR et induisent leur destruction par les macrophages du foie et de la rate, on appelle ce phénomène hémolyse extravasculaire. Quand elle est importante elle peut provoquer une anémie, un ictère et une fatigabilité. Les inhibiteurs proximaux qui empêchent la formation de ces fragments de compléments sont efficaces pour bloquer l’hémolyse intravasculaire et extravasculaire.
Retrouvez également le replay de la 6ème journée nationale Médecins/Patient (juin 2023) sur les nouveaux inhibiteurs proximaux dans l’HPN en cliquant ici.
Depuis avril 2022, le pegcetacoplan est disponible et autorisé en France, accessible dans les pharmacies de ville. Il s’agit d’un inhibiteur de la protéine C3 du complément, administré par perfusion sous-cutanée deux fois par semaine, avec possibilité d’auto-administration pour les patients formés à cet effet.
Depuis mai 2024, l‘iptacopan est disponible en accès précoce en France, accessible dans les pharmacies hospitalières avec une délivrance contrôlée patient par patient. Il s’agit d’un inhibiteur du facteur B administré par voie orale deux fois par jour. Il est très efficace mais sa durée d’action est courte nécessitant une observance parfaite. Si pour une raison ou une autre, le traitement ne peut être pris correctement, il est impératif que le patient contacte immédiatement son médecin réfèrent.
Depuis juillet 2025, le danicopan , en association avec le ravulizumab ou l’éculizumab, est également disponible en France, accessible dans les pharmacies de ville. Il s’agit d’un inhibiteur du facteur D administré par voie orale.
Comme les anti-C5, les inhibiteurs proximaux exposent au risque d’infection à méningocoque et imposent une vaccination régulière contre ces germes et la prise d’une antibioprophylaxie. Ils exposent cependant à d’autres infections justifiant la vaccination contre le pneumocoque et/ou Haemophilus influenzae avant le début du traitement et tous les 5 ans par la suite.
Pour tous les inhibiteurs du complément, la vaccination contre la grippe saisonnière est recommandée car c’est un facteur de risque d’infection à méningocoque et d’hémolyse. Les vaccins vivants atténués (ROR et fièvre jaune) sont possibles sous anti-C5 s’il n’y a pas de ciclosporine associée ; il n’y a pas de données sous inhibiteurs proximaux, raison pour laquelle ils sont officiellement contrindiqués.

Les immunosuppresseurs
Ils sont utilisés en cas d’aplasie médullaire associée mais n’ont aucun effet sur l’HPN.
Quelle que soit l’option choisie par le patient et son médecin, il est primordial de respecter les modalités de traitement mises en place. En effet, les complications liées à l’HPN peuvent se révéler très invalidantes et constituent une menace vitale. Les accidents thrombotiques en particulier peuvent endommager les organes vitaux de façon parfois irréversible.
Le suivi
Les patients atteints d’HPN traités ou non doivent avoir un suivi hématologique spécialisé qu’ils soient traités ou non. Ce suivi a pour objectif de :
- Surveiller l’hémolyse et dépister les manifestations de la maladie chez les patients non traités pour mettre en place le traitement avant la survenue de complications thrombotiques,
- Surveiller l’efficacité du traitement chez les patients traités, vérifier les vaccinations et la bonne compréhension par les patients des situations à risque nécessitant de consulter,
- Dépister la survenue d’une aplasie médullaire ou d’une maladie tumorale de la moelle, cela justifie la réalisation de myélogramme régulièrement,
- Aider les patients dans leurs démarches administratives afin de limiter au maximum le retentissement sur leur vie sociale, professionnelle et la scolarité pour les plus jeunes.
L’HPN ne nécessite aucune précaution particulière en terme d’alimentation. En dehors d’une thrombopénie associée exposant à un risque de saignement, il n’y a pas de contrindication à la pratique d’aucune activité sportive. Cette dernière est même recommandée pour limiter la désadaptation cardiovasculaire également source de fatigue chronique.
La recherche
Actuellement de nombreuses équipes, en France et dans le monde, travaillent à une meilleure compréhension des processus de l’HPN et de l’aplasie médullaire ainsi qu’à l’amélioration de leurs traitements et de leur suivi à long terme.
Au niveau de la recherche fondamentale, de nouvelles cibles pour les traitements sont en cours d’investigation.
En parallèle de nombreuses études cliniques continuent d’être menées pour l’amélioration des traitements existants, afin de toujours mieux garantir leur sécurité et leur efficacité.
Participer à un projet de recherche
Les personnes malades détiennent énormément d’informations qui sont essentielles pour mieux comprendre la maladie et trouver de meilleurs traitements. Découvrez les projets de recherche sur l’HPN en cliquant ici.
C’est grâce à la recherche, que les traitements de l’HPN (Eculizumab,Ravulizumab, Pegcetacolan, Iptacopan, Danicopan) ont pu être développés et que leur efficacité a pu être démontrée. Cela contribue au contrôle des symptômes de la maladie et à l’amélioration de la qualité de vie des patients.
HPN et l’aplasie
L’HPN et l’aplasie médullaire sont étroitement liées. En effet, une aplasie survient chez 20 à 30% des patients ayant une HPN hémolytique et l’HPN apparaît chez 30 % des patients aplasiques traités par immunosuppresseurs.
L’aplasie médullaire est également une maladie rare, dont l’incidence est de moins de 10 cas par million et par an. Elle se caractérise par une incapacité de la moelle osseuse à produire normalement les cellules sanguines (globules rouges, globules blancs et plaquette) et donc à remplacer les cellules circulantes destinées à mourir naturellement.
En fonction des types de cellules touchées, les symptômes et les risques sont différents :
- s’il s’agit de globules rouges, l’aplasie entraîne une anémie,
- s’il s’agit des globules blancs, le déficit entraîne une baisse de l’immunité et provoque ainsi des infections à répétition,
- s’il s’agit des plaquettes, cela provoque des troubles des saignements.
Sans traitement, l’aplasie médullaire est grave, mais la sévérité des symptômes varie d’un patient à l’autre.
La transfusion est le traitement de support le plus courant, elle permet de pallier au manque des cellules sanguines, les antibiotiques sont également nécessaires en cas d’infection liée à la baisse des globules blancs.
Le traitement de l’aplasie médullaire acquise repose sur la greffe de moelle osseuse ou un traitement immunosuppresseur en fonction de l’âge du patient et de la disponibilité d’un donneur compatible. Le choix du traitement n’est pas impacté par l’existence d’une HPN associée.
Documentations patient

HPN-HoPE : les patients au cœur de l’éducation thérapeutique en vidéo
L'association HPN France – Aplasie Médullaire a contribué à la création du programme HPN-HoPE, un dispositif d'éducation thérapeutique destiné aux personnes atteintes d'hémoglobinurie paroxystique nocturne (HPN). Construit avec une équipe pluridisciplinaire et en...

Le lien caché : Aplasie Médullaire et HPN
Dans le cadre de sa série de vidéos illustrées sous forme d’enquête, notre association publie aujourd’hui un nouvel épisode consacré au lien entre l’aplasie médullaire et l’HPN (Hémoglobinurie Paroxystique Nocturne). Ces deux maladies rares du sang sont étroitement...

HPN: Enquête sur une maladie
Dans le cadre de sa série de vidéos illustrées sous forme d’enquête, notre association publie aujourd’hui un nouvel épisode consacré à l’HPN (Hémoglobinurie Paroxystique Nocturne). Cette série pédagogique a pour objectif de mieux faire comprendre l’HPN, l’aplasie...
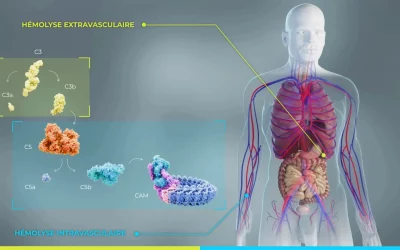
HPN : pourquoi les globules rouges sont détruits ? comprendre l’hémolyse intravasculaire (HIV) et extravasculaire (HEV)
Dans l’hémoglobinurie paroxystique nocturne (HPN), une mutation acquise du gène PIGA empêche les globules rouges de se protéger correctement contre le système du complément, un mécanisme naturel de défense de l’organisme. Privés de leurs protéines protectrices (CD55...

Parcours de soins et vécu : ce que disent vraiment les patients
La maladie rare ne se résume pas à un diagnostic, à des résultats biologiques ou à un protocole de traitement. Elle s’inscrit dans le quotidien, dans le corps, dans la tête, dans la vie sociale et professionnelle.C’est cette réalité-là que les patients atteints...

Comprendre l’HPN avec le Pr. Régis Peffault de Latour
Le Professeur Régis Peffault de Latour, coordinateur du Centre de Référence des Aplasies Médullaires Acquises et Constitutionnelles (CeRAMIC), nous éclaire sur l’HPN (Hémoglobinurie Paroxystique Nocturne), une maladie rare du sang. Dans cette vidéo, il aborde...
Publications médicales
Livret d’information pour les patients atteints de HPN
Notre Association HPN France-Aplasie Médullaire a participé activement à l’élaboration du livret « Prise en charge d’une hémoglobinurie paroxystique nocturne », publié au mois d’Octobre 2022, par le Centre de Référence des Aplasies Médullaires acquises et constitutionnelles. Vous y trouverez de nombreuses réponses aux questions que vous vous posez sur cette pathologie et sur ses conséquences.
Pour le consulter, cliquez ici.
Le suivi d’un patient HPN sous eculizumab et Ravulziumab
Le centre de référence des Aplasies Médulaires a publié un fascicule, destiné aux médecins, pour les guider et les conseiller dans le cadre du suivi d’un patient HPN traité sous eculizumab.
Pour le consulter, c’est ici.
AUTRES PUBLICATIONS
Mise à jour du PNDS (Protocole National de Diagnostic et de Soins) sur les Aplasies Médullaires Acquises et Constitutionnelles (et l’HPN) – Mai 2023
Notre Association a participé à la mise à jour du PNDS, publié en Mai 2023, par le Centre de Référence des Aplasies Médullaires.
Pour lire ce long et précieux document, cliquer ici
Une nouvelle approche dans le traitement de l’Hémoglobinurie Paroxystique Nocturne : Étude de l’Iptacopan – Mars 2024.
Les équipes du centre de référence des aplasies médullaires acquises et constitutionnelles de l’hôpital Saint-Louis AP-HP et de l’université Paris Cité, coordonnées par le Pr Régis Peffault de Latour, ont évalué un nouvel inhibiteur oral de la voie proximale du complément (Iptacopan, anti-facteur B, Laboratoire Novartis), dans le traitement de l’hémoglobinurie paroxystique nocturne. Les résultats de cette recherche, publiés le 14 mars 2024 dans le New England Journal of Medicine, offrent une nouvelle perspective dans la prise en charge thérapeutique de cette maladie rare. L’article complet est disponible sur le site de la Filière MaRIH, cliquer ici.
Deux études internationales ouvrent de nouvelles perspectives dans la prise en charge thérapeutique de l’HPN – Mars 2021.
Elles portent sur un anti C3 du laboratoire Apellis, administré par voie sous-cutanée, et sur un anti facteur D du laboratoire Novartis, à prendre par voie orale.
Le centre de référence des aplasies médullaires acquises et constitutionnelles coordonné par le Pr Peffault de Latour avec le soutien du centre d’investigations cliniques de l’Hôpital Saint-Louis – AP-HP dirigé par le Pr Kiladjian (AP-HP, Université de Paris) a contribué à l’évaluation de nouveaux inhibiteurs du complément (inhibiteurs proximaux) plus puissants, dans le traitement de l’hémoglobinurie paroxystique nocturne.
Notre Association HPN France-Aplasie Médullaire, ainsi que l’Association Française de la Maladie de Fanconi, et l’association francophone de la maladie de Blackfan Diamond participent à la gouvernance du centre de référence.
Pour lire les explications du Pr Peffault de Latour, cliquer ici .
Pour lire le communiqué de presse, c’est ici .
{kind=link}
Résumé de la journée du CRMR Aplasie – Octobre 2020
Nouveaux traitements de l’HPN (Pr de Latour)
Le ravulizumab qui agit de la même manière que l’eculizumab mais à une durée d’action plus longue a obtenu un avis favorable de la commission de transparence et une commercialisation est espérée en 2021 (pas de date connue à ce jour). => Bon à savoir: le ravulizumab est effectivement commercialisé en France depuis le mois de mars 2022.
– Le crovalimab qui est un anticorps monoclonal ciblant le C5 est en cours d’évaluation en France.
Pour lire les informations détaillées sur tous ces points cliquez ici.
Fiche « Focus Handicap » – Octobre 2019
Le texte « Focus Handicap » sur l’HPN a été mis en ligne sur le site d’Orphanet. Cette fiche rassemble des informations susceptibles d’aider les professionnels du handicap dans leur travail d’évaluation et d’accompagnement des personnes atteintes de cette pathologie. Elle a été réalisée en collaboration avec le Dr Sicre de Fontbrune, du Centre de Référence des Aplasies Médullaires acquises et constitutionnelles à l’Hôpital Saint Louis, et avec notre Association HPN-AM.
Pour consulter ce texte, cliquer ici.
Nouveaux traitements pour l’HPN – Février 2019
Bonne nouvelle: La recherche avance pour le traitement de l’HPN! Suite à la publication des résultats positifs obtenus avec le Ravulizumab (Eculizumab longue vie), notre Association a interrogé le Professeur Régis Peffault de Latour sur les nouveaux médicaments attendus pour le traitement de l’HPN. Sa réponse montre que plusieurs laboratoires s’intéressent à cette pathologie et que la recherche est en pleine effervescence.
Paris, le 5 Février 2019
Les essais concernant l’Eculizumab longue vie (étude 301) sont publiés début février dans la revue Blood qui est une très bonne revue d’hématologie.
Le premier essai qui consistait à traiter les patients toutes les deux semaines, naïfs de tout traitement avec une hémolyse dans le cadre d’une HPN, a montré que le traitement marchait aussi bien que l’Eculizumab.
Le deuxième essai qui consistait à traiter des patients déjà sous Eculizumab par le traitement par Ravulizumab (Eculizumab longue vie), s’est avéré lui aussi, non inférieur à l’Eculizumab.
Les deux essais sont donc désormais publiés. La FDA qui est l’autorité américaine des médicaments, a déjà approuvé le traitement qui est maintenant commercialisé aux Etats Unis. Les demandes sont en cours en Europe et nous pouvons espérer que cette autorisation soit disponible au mieux avant l’été et en tout cas, en 2019, en France. Les essais continuent avec l’arrivée du Ravulizumab (Eculizumab longue vie) par voie sous-cutanée avec un début des inclusions dans les protocoles, prévu mi-février.
Dans le même temps, d’autres protocoles sont ouverts ou à l’étude dans le cadre de l’hémoglobinurie paroxystique nocturne hémolytique à savoir :
– Le laboratoire Roche qui développe un traitement (Phase II) lui aussi dirigé contre le C5 par voie sous-cutanée, qui est en cours et qui va inclure des patients non contrôlés par Eculizumab.
– Le laboratoire Novartis qui développe un protocole (Phase II) de traitement par voie orale, contre la voie alterne du complément, pour les patients qui sont toujours hémolytiques sous Soliris en plus du Soliris.
– Le protocole Apellis (Phase III) qui devrait arriver probablement fin février et qui a été retardé par des problèmes de disponibilité de drogue qui lui, bloque la voie classique du complément de manière plus proximale (antiC3).
Cartes d’urgences pour l’HPN – Juin 2018
Lors du congrès des urgences de juin 2018, la filière de santé des maladies rares immuno-hématologiques MaRIH a pu présenter, en compagnie des autres filières, les nouvelles cartes d’urgence maladies rares.
Ce document est destiné aux soignants aussi bien qu’aux patients, il informe les conduites à tenir en cas d’urgence. Il doit être complété par les médecins en charge du patient (Carte HPN pour des patients hors Soliris).
Les cartes sont désormais disponibles dans les centres de Reference MaRIH.
Voir la carte d’urgence pour l’HPN hors Soliris en cliquant ici
Avancée majeure dans le traitement de patientes enceintes et atteintes d’Hémoglobinurie Paroxystique Nocturne (HPN) – Septembre 2015
Des chercheurs français viennent d’accomplir une avancée considérable pour le traitement de patientes enceintes et atteintes d’Hémoglobinurie Paroxystique Nocturne (HPN). Cette maladie rare des cellules du sang peut entraîner de graves complications pouvant aller jusqu’au décès, notamment en cas de grossesse.
Les résultats viennent d’être publiés dans la prestigieuse revue New England Journal of Medicine du 10 septembre 2015 par le Professeur Régis Peffault de Latour, du centre de référence aplasie médullaire de l’hôpital Saint-Louis (Assistance Publique-Hôpitaux de Pariset université Paris Diderot, Sorbonne Paris Cité) (http://www.aplasiemedullaire.com/) en collaboration avec le Docteur Richard Kelly de l’hôpital de Leeds (Grande-Bretagne).
Les patientes atteintes d’HPN ne pouvaient jusqu’alors être enceintes, en raison d’un taux de mortalité maternelle et fœtale inacceptable. L’eculizumab (Soliris, Alexion pharmaceutical),anticorps qui bloque un acteur majeur du système immunitaire, a révolutionné la prise en charge de cette maladie, permettant aux patients de retrouver une espérance de vie similaire à celles des personnes du même âge non malades.
Dans l’étude dont les résultats sont publiés aujourd’hui, aucun décès n’a été observé chez les 61 patientes traitées et les complications fœtales rapportées sont comparables à celles de la population générale. De plus, la plupart des enfants ont été suivis au moins deux ans après leur naissance et ne présentent aucun problème de développement. Ce travail a été réalisé sur 75 grossesses chez 61 patientes atteintes d’HPN, traitées par Eculizumab.
Cette étude est le fruit d’une collaboration internationale qui a permis en peu de temps de recueillir des informations détaillées sur un grand nombre de grossesses survenant chez des patientes avec HPN traitées par Eculizumab dans le monde. Cette avancée importante illustre aussi la qualité de la prise en charge des maladies rares en France au sein de centres de référence dédiés, et plus récemment de filières de santé, qui permettent à chaque patient derecevoir rapidement un traitement adapté. Le centre de référence aplasie médullaire appartient d’ailleurs à la filière Maladies rares Immuno-Hématologiques (MaRIH), basée à l’hôpital Saint-Louis (APHP) (http://www.marih.fr).